
ITSxpress: a CLI and QIIME 2 plugin for ITS trimming
Collaborators
- Terrence Gardener, NCSU, Raleigh, NC
Project
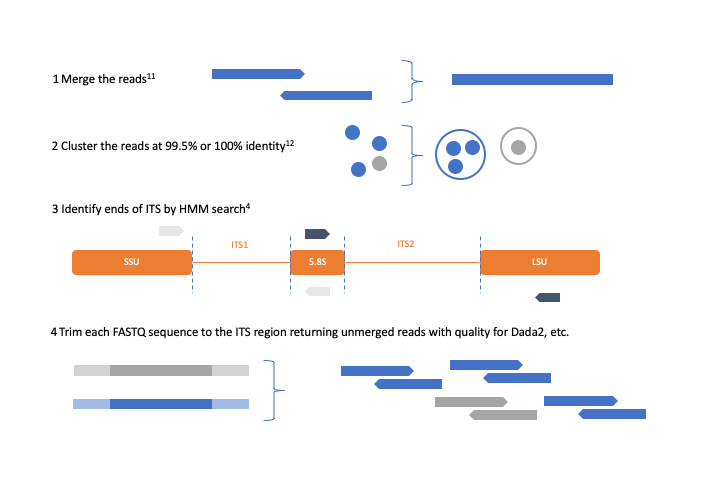
The internally transcribed spacer (ITS) region between the small subunit ribosomal RNA gene and large subunit ribosomal RNA gene is a widely used phylogenetic marker for fungi and other taxa. The eukaryotic ITS contains the conserved 5.8S rRNA and is divided into the ITS1 and ITS2 hypervariable regions. These regions are variable in length and are amplified using primers complementary to the conserved regions of their flanking genes. Previous work has shown that removing the conserved regions results in more accurate taxonomic classification1. An existing software program, ITSx, is capable of trimming FASTA sequences by matching hidden Markov model profiles to the ends of the conserved genes using the software suite HMMER.
ITSxpress was developed to extend this technique from marker gene studies using Operational Taxonomic Units (OTU’s) to studies using exact sequence variants; a method used by the software packages Dada2, Deblur, QIIME2, and Unoise. The sequence variant approach uses the quality scores of each read to identify sequences that are statistically likely to represent real sequences. ITSxpress enables this by processing FASTQ rather than FASTA files. The software also speeds up the trimming of reads by a factor of 14-23 times on a 4-core computer by temporarily clustering highly similar sequences that are common in amplicon data and utilizing optimized parameters for Hmmsearch. ITSxpress is available as a QIIME 2 plugin and a stand-alone application installable from the Python package index, Bioconda, and Github
Publications
Bolyen, E., Rideout, J.R., Dillon, M.R., Bokulich, N.A., Abnet, C.C., Al-Ghalith, G.A., Alexander, H., Alm, E.J., Arumugam, M., Asnicar, F., Bai, Y., Bisanz, J.E., Bittinger, K., Brejnrod, A., Brislawn, C.J., Brown, C.T., Callahan, B.J., Caraballo-Rodríguez, A.M., Chase, J., Cope, E.K., Da Silva, R., Diener, C., Dorrestein, P.C., Douglas, G.M., Durall, D.M., Duvallet, C., Edwardson, C.F., Ernst, M., Estaki, M., Fouquier, J., Gauglitz, J.M., Gibbons, S.M., Gibson, D.L., Gonzalez, A., Gorlick, K., Guo, J., Hillmann, B., Holmes, S., Holste, H., Huttenhower, C., Huttley, G.A., Janssen, S., Jarmusch, A.K., Jiang, L., Kaehler, B.D., Kang, K. Bin, Keefe, C.R., Keim, P., Kelley, S.T., Knights, D., Koester, I., Kosciolek, T., Kreps, J., Langille, M.G.I., Lee, J., Ley, R., Liu, Y.-X., Loftfield, E., Lozupone, C., Maher, M., Marotz, C., Martin, B.D., McDonald, D., McIver, L.J., Melnik, A. V., Metcalf, J.L., Morgan, S.C., Morton, J.T., Naimey, A.T., Navas-Molina, J.A., Nothias, L.F., Orchanian, S.B., Pearson, T., Peoples, S.L., Petras, D., Preuss, M.L., Pruesse, E., Rasmussen, L.B., Rivers, A., Robeson, M.S., Rosenthal, P., Segata, N., Shaffer, M., Shiffer, A., Sinha, R., Song, S.J., Spear, J.R., Swafford, A.D., Thompson, L.R., Torres, P.J., Trinh, P., Tripathi, A., Turnbaugh, P.J., Ul-Hasan, S., van der Hooft, J.J.J., Vargas, F., Vázquez-Baeza, Y., Vogtmann, E., von Hippel, M., Walters, W., Wan, Y., Wang, M., Warren, J., Weber, K.C., Williamson, C.H.D., Willis, A.D., Xu, Z.Z., Zaneveld, J.R., Zhang, Y., Zhu, Q., Knight, R., Caporaso, J.G., 2019. Reproducible, interactive, scalable and extensible microbiome data science using QIIME 2. Nat. Biotechnol. 37, 852–857. doi:10.1038/s41587-019-0209-9
Rivers, A.R., Weber, K.C., Gardner, T.G., Liu, S., Armstrong, S.D., 2018. ITSxpress: Software to rapidly trim internally transcribed spacer sequences with quality scores for marker gene analysis. F1000Research 7, 1418. doi:10.12688/f1000research.15704.1